BLAST Sequence
Using BLAST to compare sequence similiarity is a very frequent requests when analyzing, thus a BLAST function was integrated on BLAST tab under Tools category.
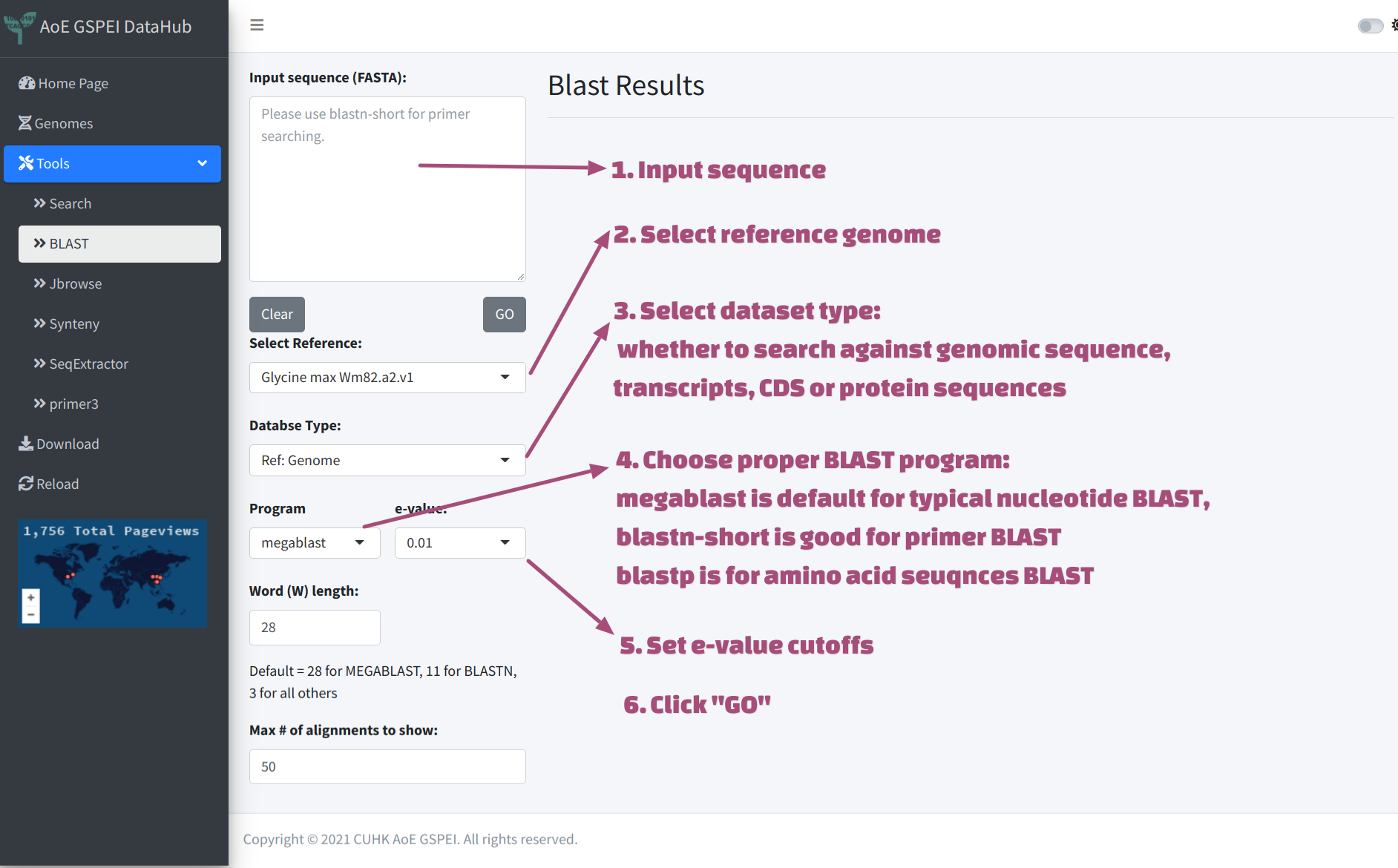
After inputting sequence, users will have to:
- choose Reference genome from drop-down menu
- select DataSet Type to search against different resource:
- genomic sequences
- transcript cDNA sequences
- transcript CDS sequences
- protein amino acid sequences
- Choose proper BLAST program, the description of the programs
could be referred to BLAST’s tasks
and blastn application options from
NCBI’s book BLAST Command Line Application User Manual. Quoted below:
- “megablast” task is optimized for intraspecies comparison as it uses a large word-size
- “blastn” is better suited for interspecies comparisons with a shorter word-size
- “tblastn” for a standard protein-translated nucleotide comparison
- “blastn-short” optimized for sequences less than 30 nucleotides
- Set e-value cutoff, 1e-10 is good to go, the default one is 0.01, which is more relaxed
- click “GO”
The BLAST result will be returned on the right panel. The database information will be shown on the top. The result table will summarize subject length, identity, alignment length, bitscore, and e-value for all the hits. User could click any hit for alignment detail. The search widget could help user to confirm if any subject of interest is in the result table. User could also click the small triangle aside of column name to sort the table based on column.

Detail of alignment will be shown under the summary table, with the information including number of matched and gap nucleotides.
